System Validation
System validation
The necessity of validation of computerized systems should follow the classification according to GAMP 5 (ed. ISPE 2008):
Category 1
Infrastructure
Operating systems, programming languages, table calculations, statistics packages, etc.
Identification, evidence of correct installation.
Category 2
Cancelled since GAMP 5
—
—
Category 3
Non configurable products
Software for measuring instruments, parametrisation possible, no adjustment to business processes
URS, risk-based vendor audit, evidence of correct installation, risk-based tests against specification according to instructions, use as intended.
Category 4
Configurable products
LIMS, data capturing software, building management systems, monitoring systems, table calculation etc.
Configuration for business processes
URS, risk-based vendor audit, evidence that the client has a QM-System, evidence of correct installation, risk-based testing to demonstrate application works as designed within the business process, procedures in place for maintaining compliance and fitness for intended use.
Category 5
Client-specific uses
Individually developed software, client-specific logic of SPS, table calculation with makros
As Category 4 with more rigorous supplier assessment, design and source code review.
Central part of every validation procedure is the Validation Plan as approved by the process owner. It is of decisive relevance for the quality of the system and the acceptance in inspections and audits (see R. Kobelt, D. Wachtmann, P.M. Kaiser & V. El-Samalouti (2001) Audit 2000. VI. Validierung von Computersystemen in der klinischen Prüfung, Pharm.Ind. 63, No. 5, 439-443 [Part I], and ibid., No. 6, 571-576 [Part II]).
The Validation Plan (VP) determines the entire process of the particular steps during the validation. If there are extensive of a series of systems to bevalidated a Validation Master Plan should be preapred (VMP). One level below particular VPs will demonstrate the partly systems.
The validation strategy should follow the V model which is graphically presented as follows:
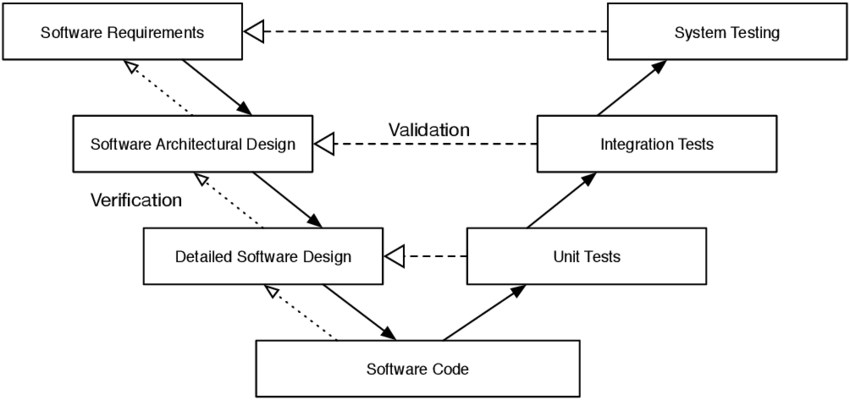
The particular steps of the V Model start with the user requirements, then go through the design of the software to be developed (left, descending wing of the V) to programming (program coding) (top at the base of the V), then to the tests (ascending right wing of the V) to the accepted software. What is missing here is the continuation into the productive phase: „maintenance“ (maintenance, further development) and „change control“ (controlled, traceable changes of hardware and software (e.g., changes of the operating system alone, the documentation, etc.; after that, if necessary, it has to be validated again).
Note:
The V Model is a logical model, no physical. This means: not all steps in the V model must be passed through identically for a certain system (e.g., purchased systems must not be validated; they are socalled „Off-The-Shelf“ systems). However, the installation procedure (IQ) is to be documented.
[DQ: Design Qualification; IQ: Installation Qualification; OQ: Operational Qualification; PQ: Performance Qualification][See also R.Kobelt, P.M.Kaiser & D. Wachtmann (2002) Spezifische Aspekte der Validierung bei klinischen Studien, die mit Hilfe eines ”Electronic Data Capture (EDC)” Systems durchgeführt werden. Am Beispiel von DATATRAK EDC™, Pharm.Ind. 64, Nr. 2, 120-127 and P.M. Kaiser, P.-H. Sanden & T. Badautschek (2007) Validation of Computer Systems in Clinical Trials, in GCP Auditing. Methods and Experiences, ed. by DGGF e.V., Aulendorf (seconed edition)]
At the end of the day validation of processes or system should be audited; we use our own developed check lists for CSV audits.
Further ancillary chapters of the validation plan are:
- List of terms and definitions (Glossary)
- Description of interfaces
- Risk analysis of processes and Traceability Matrix
We offer:
– Preparation of Handbooks for Computerized Systems (CS) according to requirements of the BSI (Bundesamt für Sicherheit in der Informationstechnologie) and FDA Title 21 CFR Part 11
– SOP development for CS: DQ, IQ, OQ, PQ
– Preparation of Validation Master Plans (VMP)
– Consulting in Validation Projects
– Audits of validatons (document review, process audit, SOP System Audit, Test procedure Audit, DQ/IQ Audit, OQ/PQ Audit, UAT Audit, System audit 21 CFR Part 11 with special check list etc.)
– Cryptography consulting: encryption technics, requirements of the European Directive No. 910/2014 of 23 July 2014 (in Germany: „Signaturgesetz (SigG and 1. SigÄndG) of 04 JAN 2005, Signaturverordnung (SigV)) and for USA: FDA Title 21 CFR Part 11.
Computer Systems: Installation, Testing and Validation
Handbook and/or SOPs for computerized systems (CS) were prepared according to the following scheme:
0 User Requirements Specification and/or Software Requirements Specification („Pflichten- und Lastenheft“)
1 System Design (Design Qualification/Specification, general; SDLC)
2 Responsibilities
2.1 Overall: Company Executive Board
2.2 Administration: Manager of Systems
3 Project Initiation
3.1 Project Definition: Project Manager
3.2 Budget and Resources
4 Validation and Tests
4.0 Validation Master Plan
4.1 Responsibilities
4.2 System Design Specification (DQ)
4.3 Risk Analysis
4.4 System Setup/Installation (IQ)
4.5 System Operation (OQ)
4.6 System Performance (PQ)
4.7 System Maintenance
4.8 Data Backup, Recovery, and Contingency Plans
4.9 Security
4.10 Validation Summary Report
4.11 QA: Audit of Validation
4.12 Change Control
5 Release of the System
6 Training Plan (for users), User Manual
7 Disposal of Project